
40 Sickle Cell (Hemoglobin SS) Disease
Michelle To and Valentin Villatoro
-
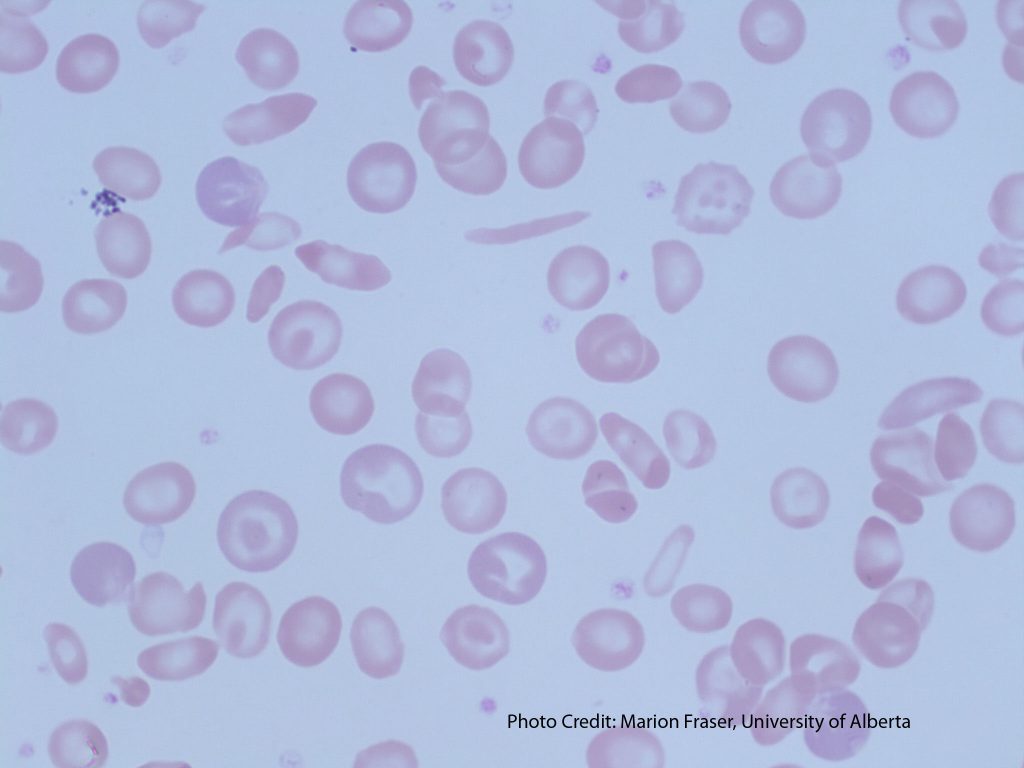
- An image of a peripheral blood smear of a patient with sickle cell disease. From MLS Collection, University of Alberta, https://doi.org/10.7939/R3G737K46
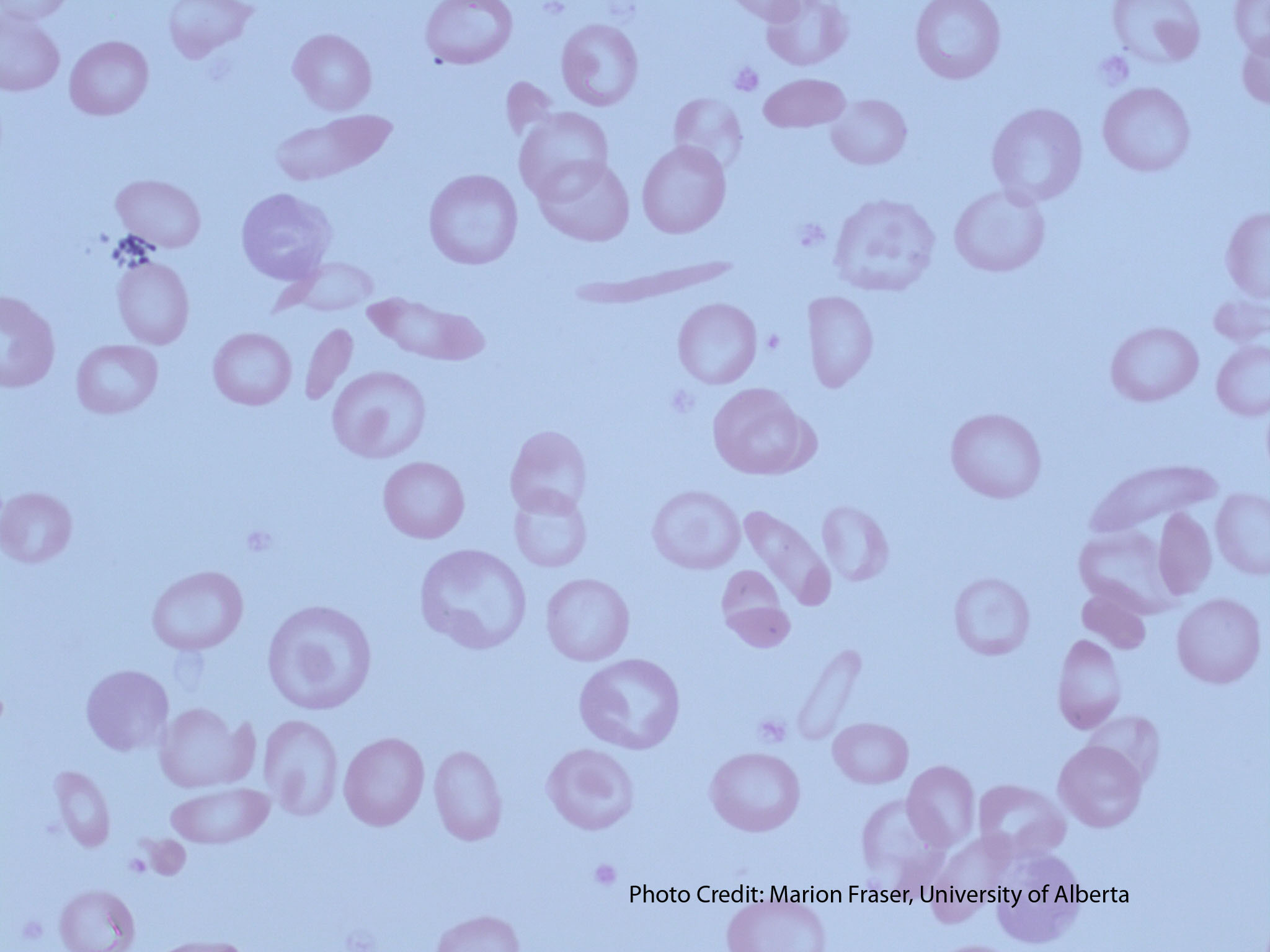
-
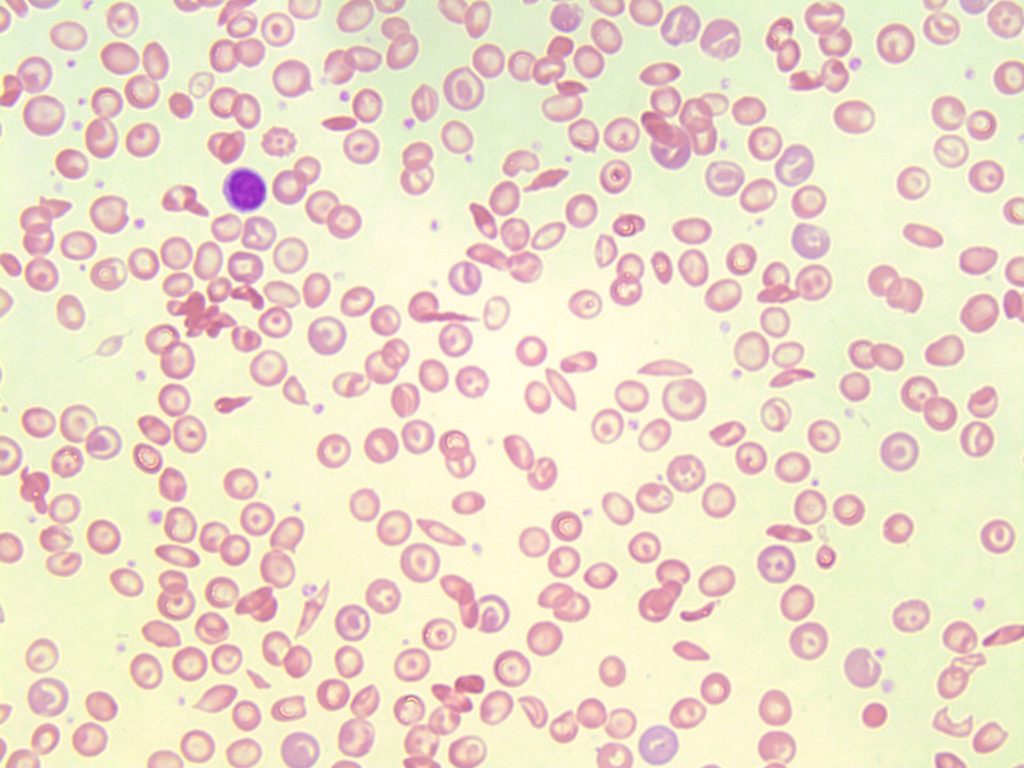
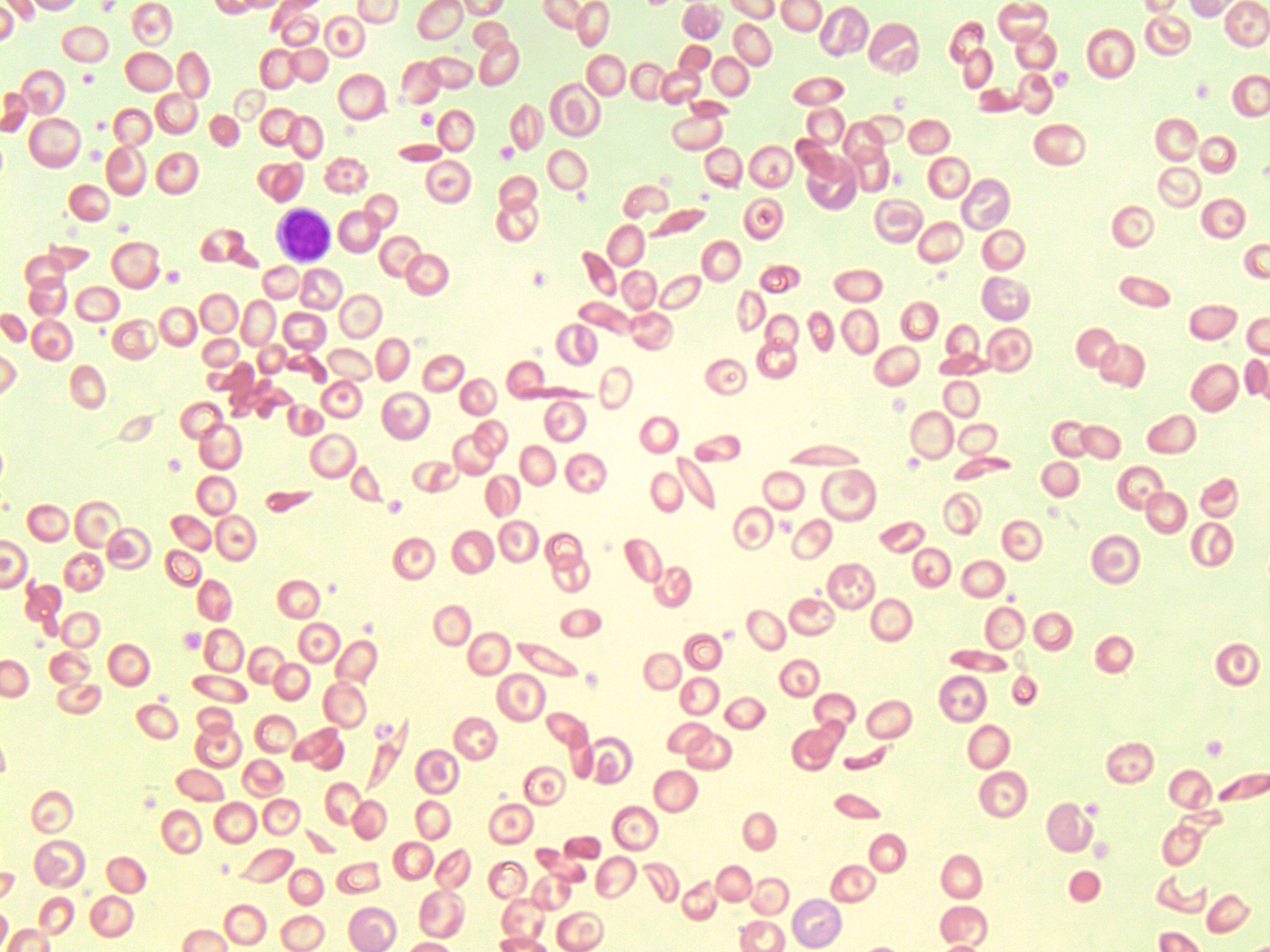
- An image of a peripheral blood smear containing sickle cells, target cells, and increased polychromasia. 50x oil immersion. From MLS Collection, University of Alberta, https://doi.org/10.7939/R3XP6VJ98
Cause(s):
β globin chain amino acid substitution in the 6th position from glutamic acid (Glu) to valine (Val). In the homozygous form of the disease, both β globin genes are affected.1
Inheritance:
Autosomal dominant2
Demographics:3
Tropical Africa
Mediterranean Areas
Sickle cell disease is common in areas where malaria is prominent and it is suggested that the disease acts as a protective factor for malaria. This protection is only seen in heterozygotes, as homozygotes often lose splenic function, which is essential for combating the parasite.
Cellular Features:1-4
See sickle cell (drepanocytes) under RBC morphology for more information about cell formation.
The formation of sickle cells becomes irreversible over time leading to the formation of rigid and “sticky” sickle cell aggregates resulting in many complications.
Complications:1-4
Chronic hemolytic anemia
Vaso-occlusion (can lead to ischemic tissue injury, splenic sequestration of RBCs, autosplenectomy)
Prone to infections
Nephropathies
Stroke
Laboratory Features of Sickle Cell Disease:2-4
|
CBC: RBC: Decreased WBC: Increased PLT: Increased Hb: Decreased RETIC: Increased RDW: Increased |
PBS: Sickle cells Normochromic, normocytic RBCs Target cells Polychromasia nRBCs Howell-Jolly bodies Pappenheimer bodies Basophilic Stippling |
BM: Erythroid Hyperplasia Iron stores: often increased |
|
Hemoglobin Electrophoresis: Hb S: 80-95% Hb A: None Hb A2: 2-% Hb F: 5-20% |
Other Tests: Solubility Screen: Positive Metasulfite Sickling Test: Positive HPLC Hemoglobin Electrophoresis |
References:
1. Chonat S, Quinn CT. Current standards of care and long term outcomes for thalassemia and sickle cell disease. Adv Exp Med Biol [Internet]. 2017 [cited 2018 Jun 5];1013:59–87. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC5720159/
1. Randolph TR. Hemoglobinopathies (structural defects in hemoglobin). In: Rodak’s hematology clinical applications and principles. 5th ed. St. Louis, Missouri: Saunders; 2015. p. 426-453.
3. Laudicina RJ. Hemoglobinopathies: qualitative defects. In: Clinical laboratory hematology. 3rd ed. New Jersey: Pearson; 2015. p.231–50.
4. Harmening DM, Yang D, Zeringer H. Hemolytic anemias: extracorpuscular defects. 5th ed. Philadelphia: F.A. Davis Company; 2009. p. 250-79).
